
Administración Nacional de Medicamentos, Alimentos y Tecnología Médica
ESPECIALIDADES MEDICINALES
Disposición 5040/2006
Apruébase el Régimen de Buenas Prácticas para la Realización de Estudios de Biodisponibilidad/ Bioequivalencia.
Bs. As., 6/9/2006
VISTO la Ley Nº 16.463, la Ley 24.766, el Decreto PEN Nº 150/92, la Resolución Secretarial Nº 46/ 2003, las Disposiciones ANMAT Nº 5330/97, 3185/99, 2807/02, 3598/02, 4290/02, 5318/02 y 7062/02, el Expediente Nº 1-47-17258-06-1 del registro de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica, y
CONSIDERANDO:
Que las precitadas normas y las disposiciones complementarias dictadas en su consecuencia, constituyen el ordenamiento legal aplicable a la aprobación, registro y autorización de venta de las especialidades medicinales cuya elaboración, importación y comercialización en el país se desarrolla al amparo de los preceptos generales establecidos por la ley 16.463.
Que la aplicación de las normativas aludidas tiene como finalidad última la protección de la salud de la población mediante la adopción de un modelo fiscalizador de gestión que, sin perjuicio de la lectura objetiva de la información calificada, destine los mayores esfuerzos a la verificación continua de la eficacia, seguridad y calidad de los productos que aquélla consume.
Que estando los estudios de bioequivalencia comprendidos dentro de la metodología de la farmacología clínica, los mismos deben cumplir con las Buenas Prácticas de Investigación Clínica.
Que con tal propósito se dictó la Disposición ANMAT Nº 3185/99, por la cual se aprobaron las recomendaciones técnicas para la realización de estudios de equivalencia contenidas en el Documento: "Cronograma para Exigencia de Estudios de Equivalencia entre Medicamentos de Riesgo Sanitario Significativo".
Que los estudios de biodisponibilidad y bioequivalencia se realizan mediante una metodología ética, legal y científica internacionalmente reconocida.
Que el grado de desarrollo alcanzado actualmente por el sistema fiscalizador de nuestro país incluye el diseño y presentación de protocolos de investigación en farmacología clínica, cuyos requisitos se encuentran impuestos por la Disposición ANMAT Nº 5330/97, encontrándose allí descriptos los procedimientos de buenas prácticas de investigación en estudios de farmacología clínica.
Que la experiencia recogida a través de los años en la evaluación de las tramitaciones de solicitud de aprobación y la presentación de resultados de estudios de bioequivalencia demuestra la existencia de desvíos, incumplimientos, inconsistencias y errores formales y metodológicos que han impedido en muchos casos la conclusión adecuada del proceso de evaluación.
Que por ello se debe normatizar la forma de presentación de la información y documentación para la solicitud de estudios de bioequivalencia.
Que existe consenso internacional acerca de la forma de presentar los resultados de los estudios de biodisponibilidad y bioequivalencia, en cuanto al ordenamiento de la información y la clara expresión de los resultados obtenidos.
Que dada la experiencia adquirida en la temática, se hace necesario normatizar la presentación de los resultados cuantitativos de los estudios de biodisponibilidad y bioequivalencia, especialmente en sus aspectos estadísticos.
Que se han tenido en cuenta para el dictado de la presente disposición los avances científicos ocurridos a nivel internacional y el documento publicado por la Organización Mundial de Salud (OMS) WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS, 40th. Report. WHO Technical Report Series Nº 937, Annex 7: Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability Ginebra 2006.
Que han tomado la intervención de su competencia la Dirección de Evaluación de Medicamentos, el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos.
Que se actúa en virtud de las facultades conferidas por el 1490/92 y el Decreto 197/02.
Por ello,
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1º — Apruébase el REGIMEN DE BUENAS PRACTICAS PARA LA REALIZACION DE ESTUDIOS DE BIODISPONIBILIDAD/BIOEQUIVALENCIA, que figura como Anexo I de la presente Disposición y que forma parte integrante de la misma.
Art. 2º — Apruébanse las pautas y requisitos de información y documentación para la solicitud de autorización para la realización de estudios de biodisponibilidad/bioequivalencia que figuran como Anexo II de la presente Disposición, que forma parte integrante de la misma.
Art. 3º — Establécese que la Etapa Bioanalítica de los mencionados estudios deberá realizarse de acuerdo a las pautas y requerimientos de información y documentación que figuran en el Anexo de la presente Disposición, que forma parte integrante de la misma, en los términos de la Disposición Nº 4844/05.
Art. 4º — Los resultados de los estudios de biodisponibilidad/bioequivalencia, deberán presentarse de acuerdo a las pautas establecidas en el Anexo IV de la presente Disposición, que forma parte integrante de la misma.
Art. 5º — Los titulares de especialidades medicinales deberán cumplir los requerimientos técnicos y administrativos aprobados por los Artículos 1º, 2º, 3º y 4º de la presente Disposición, en la realización de los estudios de biodisponibilidad/bioequivalencia.
Art. 6º — Los estudios de biodisponibilidad/bioequivalencia que no cumplimenten las pautas establecidas en los artículos 1º, 2º, 3º y 4º de la presente Disposición, no serán considerados válidos para la declaración de la Bioequivalencia, cualquiera sea la etapa de desarrollo de los mismos.
Art. 7º — En toda cuestión no prevista en la presente Disposición, se aplicarán supletoriamente las prescripciones contenidas en la Disposición A.N.M.A.T. Nº 5330/97 y sus normas complementarias.
Art. 8º — El incumplimiento de la presente Disposición hará pasible a los responsables de las sanciones establecidas en la Ley Nº 16.463 y el Decreto Nº 341/92.
Art. 9º — Regístrese; dése a la Dirección Nacional del Registro Oficial para su publicación en Boletín Oficial. Comuníquese a CILFA, CAEME, COOPERALA, CAPGEN, COMRA, SAFYBI, C.O.F.A., CAPROFAC. Cumplido, archívese PERMANENTE. — Manuel R. Limeres.
ANEXO I
(Anexo sustituido por art. 1° de la Disposición N° 1746/2007 de la Administración Nacional de Medicamentos, Alimentos y Tecnologías Médicas B.O. 26/3/2007. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.)
REGIMEN DE BUENAS PRACTICAS PARA LA REALIZACION DE
ESTUDIOS DE BIODISPONIBILIDAD/BIOEQUIVALENCIA 1; 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; 15; 16; 17
CONTENIDO
1. Introducción
2. Cuando los estudios de equivalencia no son necesarios
3. Cuando los estudios de equivalencia son necesarios
4. Estudios de Bioequivalencia en seres humanos
4.1 Consideraciones generales
4.1.1 Introducción
4.1.2 Principios éticos
4.1.3 Diseño del estudio
4.1.3.1 Diseños alternativos
4.1.3.2 Principios activos con vida media prolongada (> 24 hs)
4.1.3.3 Consideraciones para estudios en estado estacionario.
4.1.3.4 Medicamentos de liberación modificada.
4.2 Voluntarios
4.2.1 Número de voluntarios
4.2.2 Pérdidas y retiros
4.2.3 Selección de los voluntarios
4.2.4 Supervisión del estado de salud de los voluntarios durante el estudio.
4.2.5 Fenotipos genéticos
4.3 Estandarización del estudio
4.4 Productos en investigación
4.4.1 Selección del producto de comparación
4.4.2 (Punto derogado por art. 5° de la Disposición N° 1263/2012 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 8/3/2012)
4.5 Conducción del estudio
4.5.1 Selección de la dosis
4.5.2 Tiempos de muestreo
4.5.3 Toma de las muestras
4.5.4 Parámetros farmacocinéticos a ser valorados
4.5.5 Estudios de metabolitos
4.5.6 Utilización de estudios con alimentos para la determinación de la Bioequivalencia
4.6 Determinación del principio activo
4.7 Análisis estadístico
4.8 Criterios para la aceptación de bioequivalencia
4.9 Presentación de resultados
1. - INTRODUCCION
Un principio activo de comprobada seguridad y eficacia terapéutica ejerce su acción farmacológica alcanzando el sitio de acción (biofase), en un número apropiado de moléculas, y durante un tiempo adecuado. Esta concentración de moléculas es prácticamente imposible de medir, por lo que se subrogan las concentraciones en circulación sistémica con la presente en biofase, evidenciada por el efecto farmacológico.
La farmacocinética, entendida como el impacto que el organismo produce en el medicamento es, en un sentido cuantitativo, el estudio de la relación entre las concentraciones plasmáticas de un principio activo respecto del transcurso del tiempo. De este modo, la farmacocinética estudia la velocidad y magnitud con la cual las moléculas de una droga pasan de un compartimiento a otro en la unidad de tiempo, a través de una membrana.
Los productos multifuente, entre los que se encuentran los productos similares, que poseen el mismo principio activo que el producto de referencia, deben mantener el mismo perfil de seguridad y eficacia, cumplir las Buenas Prácticas de Manufactura, el correcto almacenamiento y distribución. Este concepto fundamenta que dos productos que contienen el mismo principio activo, y que demuestren un perfil farmacocinético similar, poseerán idénticas concentraciones a nivel de la biofase, y por lo tanto las mismas propiedades de seguridad y eficacia.
La equivalencia in-vivo estudia el comportamiento de dos formulaciones por tres tipos de ensayos clínicos: 1) estudios farmacocinéticos (estudios de bioequivalencia); 2) estudios farmacodinámicos; y 3) estudios controlados. Los estudios de biodisponibilidad y bioequivalencia investigan la liberación y absorción con parámetros de velocidad y masa, asumiendo que la disposición (distribución, metabolismo y excreción), permanecen constantes.
Para asegurar la equivalencia entre productos se requieren estudios in-vivo, existiendo excepciones a ellos, fundamentalmente basadas en la forma farmacéutica y basadas en el Sistema de Clasificación Biofarmacéutica (SCB).
Para las formulaciones de administración parenteral de compuestos altamente solubles en agua, su seguridad y eficacia están garantizadas por el cumplimiento de las Buenas Prácticas de Manufactura, los estándares de calidad y las especificaciones de la Farmacopea respectiva.
Los productos biológicos (vacunas, derivados de la sangre y plasma, productos biotecnológicos, etc.), plantean consideraciones que están fuera de los alcances de este Régimen.
Este Régimen brinda los lineamientos que la A.N.M.A.T. solicita para la realización de estudios clínicos de Biodisponibilidad/Bioequivalencia, su planificación, diseño, y presentación de los resultados.
2.- CUANDO LOS ESTUDIOS DE EQUIVALENCIA NO SON NECESARIOS.
De acuerdo a lo establecido en la Disposición ANMAT Nº 3185/99, III, Punto 2.1. y Disposición ANMAT Nº 2814/02.
3.- CUANDO LOS ESTUDIOS DE BIOEQUIVALENCIA SON NECESARIOS
De acuerdo a lo establecido en la Disposición ANMAT Nº 3185/99, III, Punto 4.
4. - ESTUDIOS DE BIOEQUIVALENCIA EN SERES HUMANOS
4.1. - Consideraciones generales
4.1.1 Introducción
Los estudios de bioequivalencia son estudios clínicos que deben ser llevados a cabo siguiendo las Buenas Prácticas Clínicas (Declaración de Helsinki, ICH, etc.).
4.1.2 Principios éticos
En toda investigación en Farmacología Clínica deberá prevalecer el bienestar individual de los sujetos sometidos a estudio, por sobre los intereses de la ciencia y de la comunidad.
La realización de ensayos de investigación en Farmacología Clínica debe llevarse a cabo con estricta observación de los principios científicos reconocidos y con escrupuloso respeto por la integridad física y psíquica de los individuos involucrados.
Los ensayos de investigación en Farmacología Clínica podrán incluir sujetos sanos o enfermos. Todas las personas intervinientes en un ensayo clínico deben estar plenamente informadas de las características del estudio, teniendo en cuenta que el estudio de bioequivalencia es un estudio noterapéutico, sin beneficio directo para el voluntario, por lo cual el mismo debe estar en condiciones de conocer las características del estudio y dar su consentimiento por escrito y firmado (Consentimiento Informado) frente a un testigo. Como todo estudio clínico, el estudio de bioequivalencia debe estar aprobado por un Comité de Etica en Investigación Clínica, preferentemente institucional e independiente del investigador y de todo otro participante en el estudio.
Los costos provenientes de la investigación clínica, serán afrontados por el patrocinante, si lo hubiere, o por el grupo investigador.
4.1.3 Diseño del estudio
El diseño, de todo estudio de bioequivalencia será estadísticamente apropiado, y justificado por el patrocinante, a fin de mantener los limites del error tipo I _ 0.05 y error tipo II = 0.20.
Los estudios de bioequivalencia se diseñan para investigar si son esencialmente similares, la velocidad y cantidad con las que se alcanza la circulación sistémica, el principio activo liberado de las formulaciones estudiadas (T y R), como manera de subrogar la equivalencia farmacodinámica (eficacia y seguridad) de ambas.
El diseño de todo estudio de bioequivalencia tenderá a reducir al máximo la variabilidad no dependiente de las formulaciones.
El diseño más habitual en bioequivalencia es un estudio de dos secuencias (TR/RT), dos períodos (Período 1/Período 2), dos tratamientos (T/R), cruzado al azar, con una dosis única en cada período, no replicado y balanceado (todos los voluntarios —en igual número en cada secuencia— deben recibir ambos tratamientos, T y R).
En caso de estudiarse más de dos formulaciones, cada voluntario debe recibir cada una de ellas.
El tiempo transcurrido entre cada dosis de Producto T y Producto R, llamado período de lavado, debe ser suficientemente largo como para que en la segunda administración ya se haya eliminado totalmente el principio activo administrado en el primer período. Este intervalo (período de lavado), deberá ser igual en todos los voluntarios y su duración será de por lo menos 5 veces la vida media terminal (vida media beta) del principio activo (por ejemplo, un principio activo con una vida media de 10 horas, determinará que el período de lavado no podrá ser menor a 50 horas). En este sentido se tendrá especial cuidado cuando el principio activo o su/s metabolito/s activo/s posean vidas medias largas (24 horas o más). En estos casos puede ser conveniente la utilización de un diseño paralelo.
Cuando exista alta variabilidad en la velocidad de eliminación entre los sujetos el período de lavado se estimará de acuerdo a la velocidad de eliminación más lenta.
En la evaluación pre-dosis, particularmente en el segundo período, no debe haber rastros de la dosis anterior o estos deben ser menores al 5% de la Concentración Máxima (Cmáx) obtenida en el primer período. Si un voluntario posee valores pre-dosis por encima del 5% de Cmáx., debe ser excluido del estudio.
En la figura siguiente se muestra el diseño clásico cruzado.
ESTUDIO CRUZADO 2 x 2
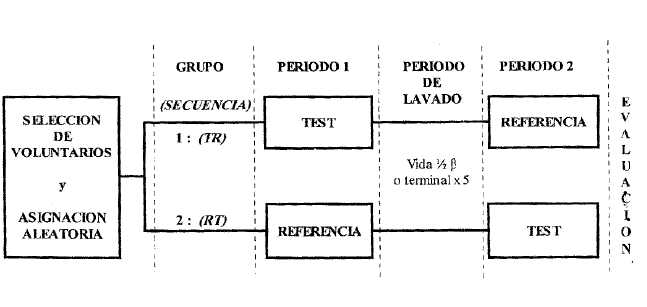
Figura 1. - Diseño clásico cruzado, no replicado
Los criterios de bioequivalencia se describen más adelante (Ver 5.8.).
4.1.3.1 Diseños alternativos
Determinados medicamentos pueden presentar efectos indeseables que no sean aceptables para los voluntarios sanos, así como requerir el empleo de dosis elevadas. En estos casos deberán utilizarse pacientes voluntarios con condición estable de su enfermedad durante todo el curso del estudio. Estas alternativas deberán ser justificadas por el patrocinante. En ciertos casos pueden requerirse estudios de diseño paralelo.
Para las llamadas drogas de "alta variabilidad farmacocinética", es decir aquellas que poseen un Coeficiente de Variación Intraindividual% igual o mayor al 30%, se recomienda que el diseño más adecuado es el diseño replicado de dos secuencias (TR / RT) y cuatro períodos (por ejemplo: Secuencia 1: RTTR; Secuencia 2 TRRT).
Todo otro diseño, como por ejemplo un diseño secuencial, deberá ser justificado por el patrocinante.
4.1.3.2 Principios activos con vida media larga (> 24 hs)
En estos casos, si el estudio cruzado resulta problemático por el excesivo período de lavado, puede recurrirse a un diseño de grupos paralelos. En estos diseños los tiempos de recolección de las muestras debe ser apropiado para permitir el tránsito gastrointestinal completo (entre 2 y 3 días). La recolección de muestras sanguíneas se extenderá hasta las 72 horas (Ver 5.5.2). Este tipo de estudios, en general, requiere mayor número de voluntarios y en algunas oportunidades, de acuerdo a las características del principio activo (razones de seguridad), deben realizarse en pacientes.
En la figura 2 se muestra un esquema de estudios con grupos paralelos.
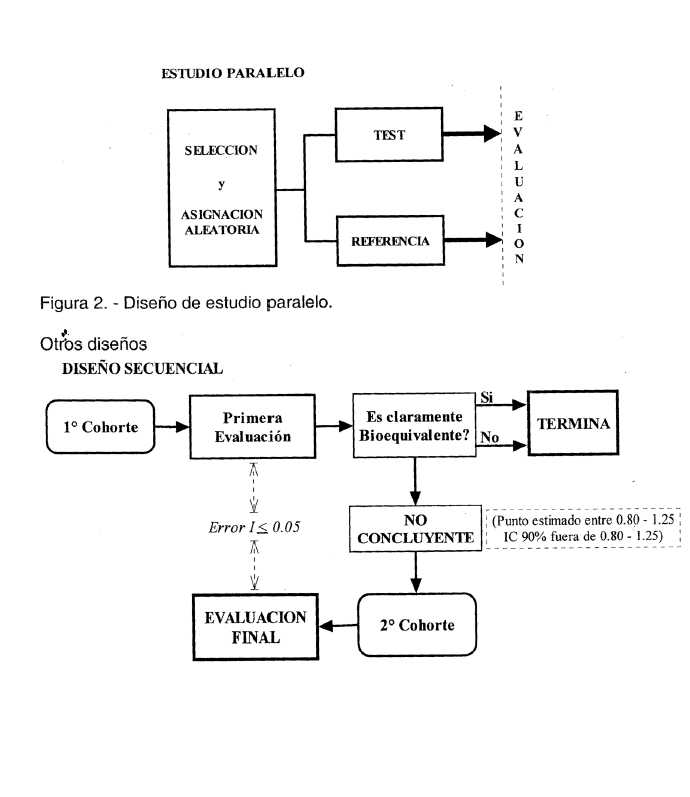
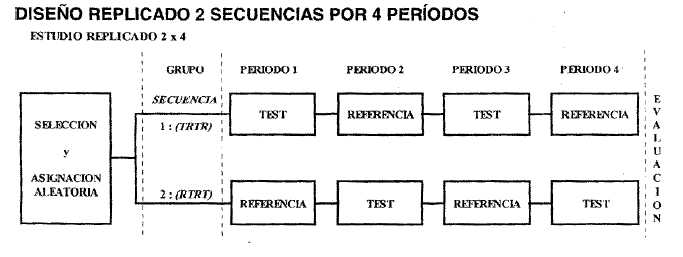
Figura 3. - Modelo de diseño replicado.
4.1.3.3 Consideraciones para estudios en estado estacionario.
Estos estudios son de utilidad cuando el principio activo es muy potente o tóxico como para ser administrado a voluntarios sanos y en dosis única. Se deberá realizar un estudio cruzado de dosis múltiples sin interrumpir la terapéutica. Los resultados pueden ser evaluados sobre parámetros farmacocinéticos o farmacodinámicos, teniendo en cuenta que estos últimos requieren, en general, un mayor número de pacientes. En estos estudios la dosificación será la establecida en el prospecto del producto de referencia y el período de lavado será no menor a tres vidas medias (tres veces el tiempo de vida media terminal).
4.1.3.4 Medicamentos de liberación modificada.
Para este tipo de medicamentos se prefieren los diseños de dosis única, a los de dosis múltiples, dada la sensibilidad de los estudios de dosis única para detectar la liberación del principio activo desde la forma farmacéutica a la circulación sistémica.
Los alimentos, es sabido, pueden modificar la liberación del principio activo desde la forma farmacéutica. En los medicamentos de liberación modificada este aspecto es crítico, por la cual los estudios de bioequivalencia con medicamentos de liberación modificada, deben realizarse con alimentos (Ver 4.3.g).
Los criterios de aceptación de bioequivalencia para productos de liberación modificada son esencialmente los mismos que para los productos de liberación simple.
4.2. Voluntarios
4.2.1. Número de voluntarios
El número de voluntarios de un estudio de bioequivalencia deberá ser calculado teniendo en cuenta la Variabilidad lntraindividual, la Máxima diferencia a ser detectada (20%; 0,20) y los errores de Tipo I (Alfa =0,05) y Tipo II (Beta=0,20).
• Se debe asignar la misma cantidad de voluntarios a cada secuencia/grupo.
• El resultado debe ser un número par, que se obtiene del redondeo superior de la estimación del tamaño de muestra.
El tamaño muestral para una razón T/R = 1, se podrá calcular de acuerdo a la siguiente fórmula propuesta por Marzo y Balant (1995):
N > 15,68 x CV intraindividual2 / ∆2
Donde:
CV Intraindividual: es el Coeficiente de Variación Intraindividual
∆2 = 0,202 = 0,04.
N: es el número total de voluntarios, N = n1 + n2; y siendo que n1 = n2.
4.2.2. Pérdidas y retiros
Se deberá reclutar un número adecuado de voluntarios, teniendo en consideración las posibles pérdidas y retiros. Las pérdidas y retiros se aceptarán si están contempladas en el protocolo y se demuestre que las mismas no afectan la potencia estadística del estudio. Los retiros de los voluntarios y sus razones (especialmente las debidas a efectos adversos), deberán constar en el informe final a ser presentado. Los voluntarios calculados "de más", serán considerados "adicionales". En el protocolo debe quedar claramente establecido si las muestras obtenidas de los voluntarios "adicionales" se analizarán aunque no se requieran para el estudio estadístico.
4.2.3. Selección de los voluntarios
En el protocolo deberán figurar claramente los criterios de selección (criterios de inclusión y criterios de exclusión). Es recomendable la inclusión de ambos sexos, no obstante dadas las características de ciertos principios activos, esta decisión será responsabilidad del patrocinante.
En el caso de drogas de alta variabilidad farmacocinética (CV% intraindividual igual o mayor al 30%) podrán acotarse los criterios de edad, sexo y peso a los efectos de establecer una mayor uniformidad en los grupos. En el protocolo deberá figurar que el investigador se asegurará que las personas de sexo femenino no se encuentren embarazadas y no se embaracen durante el curso el estudio. La confirmación que las voluntarias no están embarazadas deberá realizarse antes de la primera dosis y antes de la segunda dosis.
Los voluntarios serán mayores de edad, entre 21 y 55 años, con un peso dentro del rango de normalidad para su sexo y edad (Indice de Masa Corporal —IMC— con valores entre 19 y 27).
Los voluntarios de los estudios de bioequivalencia no deberán tener antecedentes de abuso de drogas y/o alcohol, siendo preferentemente no fumadores. Los voluntarios no deben haber sido sometidos a ningún tipo de cirugía gastrointestinal (excepto apendicectomía, no complicada, de por lo menos tres meses de antigüedad).
Cada voluntario será examinado por un médico calificado, redactando la correspondiente historia clínica y se le solicitarán estudios de laboratorio, electrocardiograma y radiografía de tórax con el objeto de asegurar la salud de los voluntarios.
Serán criterios de exclusión, entre otros, el hecho de haber sido tratado el voluntario por trastornos gastrointestinales, convulsivos, depresivos, cirugía gastrointestinal (excepto apendicectomía de por lo menos tres meses de antigüedad), alteraciones hepáticas y aquellos que presenten riesgo de alguna recurrencia de cualquier enfermedad durante el desarrollo del estudio. También serán criterios de exclusión: el haber participado en otro estudio de farmacología clínica en los últimos tres meses; el haber donado sangre dentro de los últimos tres meses; estar recibiendo cualquier tipo de medicación.
Se deberá asegurar la capacidad del voluntario para comprender y cumplir el protocolo y en consecuencia, estar en condiciones de firmar el consentimiento informado.
4.2.4. Supervisión del estado de salud de los voluntarios durante el estudio.
El estado de salud de los voluntarios, antes, durante y después del estudio, debe ser estrictamente supervisado, especialmente en lo referido a la aparición de eventos y efectos adversos, así como a la presentación de toxicidad o enfermedad/es intercurrente/es.
La incidencia, severidad y duración de las reacciones adversas que apareciesen durante el estudio clínico de bioequivalencia deberán ser comunicadas al Sistema Nacional de Farmacovigilancia, en el caso que sean graves, dentro de las 48 horas de ocurridas. Para el caso de las reacciones adversas no graves serán detalladamente informadas en el informe final.
En el protocolo del estudio deberá presentarse un modelo de formulario de registro de eventos y efectos adversos.
4.2. 5. Fenotipos genéticos
En caso de evidencia de la existencia de diferentes fenotipos genéticos, en los diseños deberá asegurarse la distribución proporcional de tales fenotipos.
4.3. Estandarización del estudio
Se deben mantener condiciones estándar en el estudio para reducir la variabilidad que no es dependiente de los productos en estudio (Producto Test y Producto de Referencia). Se entiende por condiciones estándar a la uniformidad en:
a) Dieta.
b) Ingesta de líquidos.
c) lngesta de sustancias tales como determinados jugos de fruta, alcohol, cafeína.
d) Administración de medicamentos antes y durante el estudio.
e) Postura.
f) Actividad física.
g) Especificación del momento del día del estudio en el que los voluntarios recibirán los medicamentos en estudio: los voluntarios deberán recibir los medicamentos en estudio luego de una noche con 10 horas de ayuno. La mañana del estudio los voluntarios no podrán ingerir agua en la hora anterior a la toma del medicamento. Durante la noche anterior se permitirá la ingesta de bebidas libres de cafeína y alcohol.
La dosis de cada uno de los medicamentos en estudio (Test y Referencia) será ingerida con un volumen de agua estandarizado entre 200 y 250 ml. Recién dos horas después de la toma del medicamento se permitirá la ingesta de agua.
A las cuatro horas posteriores a la toma del medicamento se permitirá la ingesta de una comida estándar, que deberá ser igual para todos los voluntarios y en ambos períodos del estudio.
Si en el prospecto del Producto de Referencia se establece que el medicamento debe ser tomado conjuntamente con los alimentos, el estudio de bioequivalencia deberá realizarse con la ingesta de alimentos. La comida de prueba deberá tener en cuenta las costumbres dietéticas locales y las características de cada medicamento. Debe ser estandarizada de acuerdo a las recomendaciones internacionales. Cuando las características del medicamento lo requieran debará poseer un alto contenido calórico. (800 a 1000 calorías), de las cuales 500 a 600 calorías provendrán de los lípidos, 250 calorías provendrán de los hidratos de carbono y 150 calorías derivarán de las proteínas.
Los medicamentos se administrarán después de no más de 30 minutos de la ingesta de la comida.
4.4. Productos en investigación
4.4.1. Selección del producto de comparación
De acuerdo a lo establecido en la Disposición ANMAT Nº 3185/99, II Definiciones.
4.4.2. (Punto derogado por art. 5° de la Disposición N° 1263/2012 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 8/3/2012)
4.5. Conducción del estudio
4.5.1. Selección de la dosis
Deberá usarse la mayor dosis comercializada en los estudios de bioequivalencia, salvo que razones de seguridad indiquen que debe utilizarse una dosis menor. Excepcionalmente, pueden utilizarse dosis más elevadas si existen problemas bioanalíticos. En este caso no se superará la dosis máxima recomendada.
Si se emplea una dosis menor, ello deberá estar justificado por razones de seguridad y estar claramente establecido en el protocolo; como puede ser el caso en que los excipientes no mantengan una proporcionalidad en su concentración, respecto al producto de mayor dosis.
4.5.2. Tiempos de muestreo
Las muestras deben tomarse con una frecuencia adecuada tal, que permita determinar fehacientemente:
• Concentración máxima (Cmáx).
• Area Bajo la Curva a tiempo t (AUC0-t).
• Area Bajo la Curva a tiempo infinito AUCinfinito (o total).
• Vida media (T½).
• Constante de eliminación terminal.
Para cumplir con este requerimiento se sugiere un mínimo de 7 a 10 muestras teniendo en cuenta las características farmacocinéticas del principio activo.
El tiempo de recolección de las muestras se halla directamente relacionado con las características farmacocinéticas del principio activo, así como de la forma farmacéutica empleada.
El investigador debe adecuar el número y distribución de extracciones con el fin de lograr la mejor detección y valoración de los parámetros farmacocinéticos estudiados.
El período de lavado no deberá ser menor a 5 vidas medias terminales del principio activo y no mayor 3 ó 4 semanas. En caso de necesitarse un período de lavado mayor se deberá considerar la posibilidad de un diseño paralelo.
4.5.3. Toma de las muestras, procesamiento, conservación y transporte (etapa previa a su bioanálisis).
En los estudios de bioequivalencia, lo habitual es tomar muestras de sangre para determinar la concentración del principio activo. En algunas oportunidades la matriz biológica es el plasma o suero. En el caso que el principio activo se elimine por la orina sin haberse metabolizado, se podrán tomar muestras de orina. Con las muestras de orina no pueden realizarse estimaciones de Tiempo en alcanzar Cmáx (tmáx) ni Cmáx.
Las muestras obtenidas de sangre, deberán ser procesadas y almacenadas bajo condiciones estandarizadas que hayan demostrado que no determinan degradación del principio que se desea evaluar. Las muestras utilizadas para control de calidad se prepararán en la matriz de interés (sangre o plasma) con concentraciones bajas, medias y altas de la curva de calibración. Estas muestras de control de calidad deben almacenarse juntamente con las muestras del estudio y analizadas con cada partida de muestras del estudio.
La metodología de recolección, procesamiento, conservación y transporte de las muestras debe estar claramente precisada y especificada en el protocolo.
4.5.4. Parámetros farmacocinéticos a ser valorados
a) En los estudios de dosis únicas, los siguientes parámetros farmacocinéticos serán calculados:
1. Area Bajo la Curva concentración/tiempo, desde tiempo 0 a tiempo t (AUC 0-t): es el área bajo la curva comprendida entre el tiempo 0 y la última determinación efectuada. Para el cálculo de esta área bajo la curva se empleará el método de integración trapezoidal, lineal o logarítmico. Esta área bajo la curva debe ser por lo menos el 80% de AUC0-Infinito, de acuerdo a las siguientes ecuaciones:
Regla Trapezoidal para cálculo de AUC:
AUC0-t = ∑(Ci+Ci-1/2) x (ti-ti-1)
Transformación logarítmica (log Trapezoidal)
∑ (Ci+Ci-1)/ (1/ ∆t)(In Ci/Ci-1)
Donde :
Ci. "Concentración mayor",
Ci-1: "Concentración menor",
ti: tiempo en obtenerse Ci,
ti-1: tiempo en obtenerse Ci-1
∆t : ti - ti-1
2.- Area Bajo la curva concentración/tiempo desde tiempo 0 a tiempo infinito (AUC 0-Infinito): es el área bajo la curva extrapolada a tiempo infinito, la cual representa la exposición total, calculada mediante la fórmula
AUC0-Infinito = AUC0-t +(Cúlt. / ke)
Donde:
Cúlt: es la última concentración mensurada.
ke: es la velocidad de eliminación terminal (calculada con un método apropiado que debe estar establecido en el protocolo).
3.- Concentración Máxima (Cmáx), es la máxima concentración obtenida, también llamada pico de concentración, que representa la exposición pico o puntual del principio activo o metabolito.
4. - Tmáx: es una medida de tiempo. Establece el tiempo transcurrido hasta alcanzar la concentración pico Cmáx.
Adicionalmente, deben calcularse:
5. - Vida media (T½): vida media terminal en la matriz biológica (sangre, plasma o suero).
6. - ke: es la constante de eliminación terminal (calculada con un método apropiado que debe estar establecido en el protocolo). Se recomienda estimarla por regresión de mínimos cuadrados de las tres últimas mediciones en fase de eliminación.
b) En los estudios en el estado estacionario, los parámetros a calcular, serán los siguientes:
1. - AUC_: Area bajo la curva en un intervalo de dosis _, en el estado estacionario.
2. - Cmáx. (Ver antes).
3. - Cmin: es la mínima concentración encontrada al final de un intervalo de dosis.
4. - Fluctuación%: (Cmáx - Cmin/ Cmin)*100. En caso de Cmin, ser muy pequeña, se empleará como denominador Cpromedio, calculada con la siguiente fórmula:
Cprom= /AUC0-t /t )
Este parámetro evalúa la fluctuación de concentración entre dos administraciones.
c) Cuando se utilicen muestras de orina, se empleará:
1. - Ae: recuperación urinaria acumulativa.
2. - Máxima velocidad de excreción (dAe / dt).
Estos parámetros reemplazan a AUC y Cmáx.
4.5.5. Estudios de metabolitos
La medición de la droga madre ("parent drug"), es más sensible para evaluar diferencias entre productos. Los metabolitos expresan más los procesos de disposición.
Se deben mensurar los metabolitos en estas situaciones:
1.- La droga en estudio es una pro-droga.
2.- Los niveles alcanzados por la droga madre son muy bajos como para establecer una exacta medición en la matriz biológica (sangre, plasma o suero).
4.5.6. Utilización de estudios con alimentos para la determinación de la Bioequivalencia
(Ver 4.1.3.4. y 4.3.g)
4.6. Determinación del principio activo
La metodología bioanalítica utilizada para la cuantificación del principio activo y/o metabolitos, debe estar bien caracterizada, validada y documentada.
La validación tiene por objeto demostrar que el método utilizado es confiable y reproducible.
En la etapa bioanalítica de los estudios de bioequivalencia, deben aplicarse las Buenas Prácticas de Laboratorio.
Se hará una presentación detallada de la metodología bioanalítica utilizada, para la determinación del principio activo, con detalle de:
a) Control de calidad o Curva Estándar de Concentración.
b) Precisión Intra-día (CV%).
c) Exactitud Intra-día.
d) Precisión Inter-día (CV%).
e) Exactitud Inter-día.
f) Linearidad.
g) Rango lineal.
h) Sensibilidad/LOQ.
i) Recuperación (%).
Es necesario conocer la estabilidad del principio activo y/o metabolito en la matriz biológica.
Toda la metodología bioanalítica debe estar detalladamente descripta en el protocolo y en el informe final.
Con cada muestra deberá estar referido según corresponda, su valor o magnitud, su unidad de medida o su característica, exclusivamente, de la siguiente manera:
• Valor obtenido del analito en la muestra extraída.
• ANM: Analito No Medible pero detectable (presencia de señal).
• AND: Analito No Detectable (ausencia de señal).
• TNE: Muestra Técnicamente No Evaluable (descartada por sus características).
• MAR: Muestra Ausente/Perdida o no obtenida según lo planeado.
4.7. Análisis estadístico
La metodología estadística deberá estar extensa y claramente expresada en el protocolo y en el informe final; estableciendo los "límites de riesgo de declarar falsamente la bioequivalencia entre dos productos. En la metodología se debe incluir la estadística descriptiva y la inferencial.
a) estadística descriptiva
1. - Información individual.
Se presentarán los datos obtenidos de cada voluntario en cada uno de los tiempos de toma de la muestra, especificando:
1.1.- Datos demográficos de la muestra (edad, sexo, peso índice de masa corporal, etc).
1.2. - Para cada individuo:
a) Unidad de medida.
b) Valores en cada tiempo.
c) Secuencia.
d) Producto recibido (T= Producto en Estudio o R= Producto de referencia).
e) Concentración máxima (Cmáx).
f) Tiempo en alcanzar Cmáx. (Tmáx).
g) Constante de eliminación (ke).
h) Vida media (T½).
i) Area bajo la curva a tiempo t (AUC0-t).
j) Area bajo la curva a infinito (AUCinf).
1.3. - Para cada concentración/tiempo:
a) Media aritmética.
b) Mediana.
c) Desvío estándar.
d) Coeficiente de Variación por ciento (CV%).
e) Valor mínimo (Mn).
f) 1º cuartilo.
g) 3º cuartilo
h) Valor máximo. (Mx).
1.4. - Curvas de cada voluntario, con datos no transformados logarítmicamente, de los valores Concentración/tiempo, recibiendo producto en estudio y producto de referencia (2 curvas por voluntario).
2. - Información consolidada
2.1. Curvas comparativas Producto en estudio y Producto de referencia con valores promedio Concentración/tiempo (datos no transformados logarítmicamente).
2.2. Tabla de la Secuencia RT, cuando cada voluntario recibió el tratamiento R (referencia) y T (producto en estudio), conteniendo para cada voluntario y cada tratamiento:
a) Cmáx.
b) Tmáx.
c) Ke.
d) T½.
e) AUC0-t.
f) AUCinf.
Para cada uno de los parámetros, expresar:
• Media aritmética (Md).
• Mediana (Mn).
• Media geométrica (MG).
• Desvío estándar.
• Coeficiente de Variación por ciento (CV%).
• Valor mínimo (Mn).
• 1º cuartilo.
• 3º cuartilo
• Valor máximo.(Mx).
2.3. Tabla de la secuencia TR, cuando cada voluntario recibió el tratamiento T y R, conteniendo para cada voluntario y cada tratamiento:
a) Cmáx.
b) Tmáx.
c) Ke.
d) T½.
e) AUC0-t.
f) AUCinf.
Para cada uno de los parámetros, expresar:
• Media aritmética (Md).
• Mediana (Mn).
• Media geométrica (MG).
• Desvío estándar.
• Coeficiente de Variación por ciento (CV%).
• Valor mínimo (Mn).
• 1º cuartilo.
• 3º cuartilo
• Valor máximo. (Mx).
En ningún caso la presencia de valores extremos o atípicos pueden ser excluidos en el análisis estadístico. Se deberá reportar y analizar estadisticamente la presencia de estos valores.
3. - Relación T/R (Punto Estimado) y su intervalo de confianza 90%.
Se expresará para cada parámetro (Cmáx, AUC0-t y AUCinf), la razón T/R (Punto Estimado) y el intervalo de confianza 90% de la misma.
b) Estadística inferencial
1. - Análisis de Variancia (ANOVA) de los datos logarítmicamente (In) transformados (Cmáx, AUC0-t y AUCInfinito).
Se presentará la tabla del ANOVA de los datos log-transformados, de cada uno de los parámetros Cmáx., AUC0-t y AUC0-Infinito, especificando las fuentes de variación (Secuencia/arrastre, Período, Tratamiento), grados de libertad, suma de cuadrados, cuadrados medios, valor del estadístico F y los valores correspondientes de p.
La Hipótesis Nula a testear con el ANOVA es:
H0: m T = m R
Tabla modelo de análisis de variancia: ver anexo III.
Coeficiente de Variación% Intraindividual:
Se debe especificará el Coeficiente de Variación Intraindividual %, obtenido éste mediante la raíz cuadrada de los cuadrados medios residuales del ANOVA, según la fórmula:

2. - Cálculo del Intervalo de Confianza 90% para la razón de medias:
Se deberá construir el Intervalo de Confianza 90% de la razón de medias (con datos logarítmicamente transformados) de los parámetros Cmáx. AUC0-t y AUC0-Infinito y luego la aplicación de dos test de hipótesis unilaterales.
3. - Resultados de las dos pruebas t unilaterales.
Se expresarán los resultados de las dos pruebas unilaterales, para cada límite del Intervalo de Confianza (Límite Inferior y Límite Superior), por ejemplo Test de Schuirmann. Las hipótesis a testear son las siguientes (H0= Hipótesis Nula; H1: Hipótesis Alterna):
H0a): LI < 0,80 H0b): LS > 1,25 (Bioinequivalencia)
Frente a:
H1a): LI ≥0,80 H0b): LS ≤1,25 (Bioequivalencia)
Para Tmáx se utilizará solamente estadística descriptiva con datos sin log-transformar. En caso de ser necesario establecer bioequivalencia para Tmáx, se emplearán métodos no paramétricos.
Para t½ y ke, se brindará solamente estadística descriptiva.
En los estudios secuenciales, la combinación de resultados de la primera y segunda etapa, se aceptará siempre y cuando se haya utilizado el mismo protocolo, los medicamentos utilizados procedan del mismo lote que el utilizado en la fase anterior y se cumpla con el "Test de Consistencia". La realización de estudios secuencia les, deberá estar prevista en el protocolo, y cuando se esperen, por las características del principio activo, resultados no concluyentes de bioequivalencia (por ej. AUC 0-t y AUC 0-Infinito, IC 90%: 0, 80-1,25 y Cmáx. IC 90%: 0, 75-1,33).
4.8. Criterios de aceptación de bioequivalencia
1. - Razón de Area Bajo la Curva (AUC0-t).
El Intervalo de Confianza 90% de esta razón debe encontrarse comprendido entre 0,80 y 1,25. El parámetro AUC0-t no deberá ser menor del 80% del parámetro AUC0-Infinito. Si la ventana terapéutica del principio activo es estrecha (menor de 2), este Intervalo de Confianza puede estrecharse, basándose en fundamentaciones clínicas de eficacia y seguridad.
2. - Razón de Cmáx
En líneas generales, también su Intervalo de Confianza 90% debe hallarse comprendido entre 0,80 y 1,25. No obstante, dada la mayor variabilidad de Cmáx; este Intervalo de Confianza puede ampliarse, fundamentándose en razones clínicas documentadas que demuestren fehacientemente que esta ampliación no produce modificaciones en la eficacia y seguridad del medicamento.
3.- Tmáx
El análisis estadístico de Tmáx se realizará si existe evidencia clínica documentada de rápido comienzo de acción del principio activo o información sobre efectos adversos relacionados. Dadas las características de Tmáx se utilizará un Intervalo de Confianza No Paramétrico, que deberá encontrarse dentro de 0, 80-1, 25.
4.9. Presentación de resultados
Los resultados deben ser presentados a la Autoridad Reguladora, de manera clara y ordenada, con el objeto de ser evaluados y que permitan el recálculo de tales resultados. Se utilizarán tablas estandarizadas por la Autoridad Reguladora (Anexo IV) en modalidad papel y electrónico bajo formato MS Excel 97 o superior.
Asimismo se deberá aportar un número adecuado de cromatogramas y (al menos los correspondientes al 20% del total de voluntarios) seleccionados al azar de manera que haya igual número de cromatogramas de voluntarios que recibieron la secuencia TR que los que recibieron la secuencia RT. Asimismo se aportarán un número representativo de formularios de consentimiento informado firmado por los voluntarios. En ambos casos la Autoridad Reguladora podrá exigir la totalidad de los cromatogramas. Para este proceso la autoridad asimismo fijará la modalidad de revisión.
El Investigador Principal debe firmar el informe final, debiendo ser también firmado por el Director Técnico del laboratorio patrocinante.
Se dejará constancia del paquete estadístico utilizado.
Deberán presentarse gráficos de las relaciones concentración/tiempo de cada voluntario habiendo recibido la formulación Test y habiendo recibido la formulación de Referencia (dos gráficos por voluntario). Estas figuras se presentarán con los datos no transformados logarítmicamente. Asimismo se presentará una figura resumen con los datos promedio (no transformados logarítmicamente) de cada tiempo ("Curvas resumen").
Se deberán presentar todos los datos, incluso los de aquellos voluntarios que hayan abandonado el estudio o representen valores extremos o atípicos.
Deberá presentarse toda la información correspondiente a los aspectos de seguridad (eventos y efectos adversos).
En el informe final deberán consignarse, tanto para el producto test como para el producto de referencia, la siguiente información:
1. — Fórmulas (referencia utilizada: Fotocopia de rótulo y prospecto)
2. — Número de lote (partida).
3. — Fecha de vencimiento.
4. — Elaborador y lugar de elaboración.
BIBLIOGRAFIA CONSULTADA
1- ANMAT (Argentina). Disposición N° 5330/97. Requerimientos de estudios de bioequivalencia.
2- ANMAT (Argentina). Disposición N° 2814/02. Formas farmacéuticas que no requieren estudios de bioequivalencia.
3- OMS. Serie de Informes Técnicos N° 863. Comité de expertos de la OMS en especificaciones para las preparaciones farmacéuticas. 34° Informe. Ginebra, 1996.
4- WHO. Multisource (Generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability. Draft revision. Working document QAS/04.093/Rev.4. 2005. www.who.int/ /medicines/services/expertcommittee/pharmprep/QAS04_093Rev 4_final.pdf
5- ANMAT (Argentina). Disposición N° 3598/02. Declaración Jurada del investigador principal.
6- Red Panamericana de Armonización de la Reglamentación Farmacéutica. Grupo de Trabajo Bioequivalencia. Criterios científicos para los ensayos de Bioequivalencia (in vivo e in vitro), las bioexenciones y las estrategias para su implementación. Documento borrador. IV Conferencia Panamericana para la Armonización de la Reglamentación Farmacéutica. República Dominicana. 2-4 Marzo 2005.
7- Declaración de Helsinki de la Asociación Médica Mundial. Principios éticos para las investigaciones médicas en seres humanos. 52a. Asamblea General. Edimburgo. Escocia, Octubre de 2000. O www.ICH.org. E6 Good Clinical Practices.
8- ANMAT (Argentina). Disposición 5330/97. Buenas Prácticas de Investigación en Farmacología Clínica.
9- Marzo, A; Balant, LP. An updated reappraisal addresed to applications of interchangeable multisource pharmaceutical products. Arzn. Forsch. Drug Res. 1995; 45(1)2: 109-15).
10- WHO Expert committee on specifications for pharmaceutical preparations. WHO Technical Report Series. Annex 11, Geneva, 2002.
11- WHO: Revision / Update of the guidance on the selection of comparator pharmaceutical products for equivalent assesment of interchangeable multisource (Generic) products. Working Document QAS/05.143/Rev. 2005. www.who.int/medicines/services/expertcommittees/pharmprep.
12- Zanen, P. Bioequivalence and generic medicines. www.egagenerics.com/doczanenbiogenerics. pdf. Fecha de acceso 29.09.05.
13- EMEA 2001 Report CPMP/1678/00 Committee for Proprietary Medicinal Products European Public Assessment Report (EPAR) Active Substance.
14- Amidon, GJ; Lennernas, H; Shah, VP; Crison, JR. A theoretical basis for a biopharmaceutical drug classification: The correlation of in vitro drug product dissolution and in vivo Bioavailiability. Pharm. Res. 1995; 12(3): 413-420.
15- Farmacopea Nacional Argentina. VII Edición. Documento borrador del Grupo de Trabajo de Bioequivalencia. Bs. As. 2005.
16- Red Panamericana para la Armonización de la Reglamentación Farmacéutica. Grupo de Trabajo en Bioequivalencia. Marco para la ejecución de los requisitos de equivalencia para los productos farmacéuticos. Agosto, 2006.
17- WHO EXPERT COMMITTEE ON SPECIFICATIONS FOR PHARMACEUTICAL PREPARATIONS, 40th. Report. WHO Technical Report Series N° 937, Annex 7: Multisource (generic) pharmaceutical products: guidelines on registration requirements to establish interchangeability Geneva 2006.
ANEXO II
(Anexo derogado por art. 1° de la Disposición N° 4010/2017 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 2/5/2017. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.)
ANEXO III
(Anexo derogado por art. 1° de la Disposición N° 4010/2017 de la Administración Nacional de Medicamentos, Alimentos y Tecnología Médica B.O. 2/5/2017. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.)
ANEXO IV
(Anexo derogado por art. 1° de la Disposición N° 4010/2017
de la Administración Nacional de Medicamentos, Alimentos y Tecnología
Médica B.O. 2/5/2017. Vigencia: a partir del día siguiente al de su
publicación en el Boletín Oficial.)
- Nota Infoleg: por art. 1° de la Disposición N° 6338/2006 de la Administración Nacional de Medicamentos, Alimentos y Tecnologías Médicas B.O. 25/10/2006 se suspenden los efectos de la presente Disposición, por el término de ciento ochenta (180) días, contados a partir del día siguiente al de su publicación en el Boletín Oficial. Abrogada por art. 2° de la Disposición N° 1746/2007 de la Administración Nacional de Medicamentos, Alimentos y Tecnologías Médicas B.O. 26/3/2007. Vigencia: a partir del día siguiente al de su publicación en el Boletín Oficial.